Медицинский эксперт статьи

Новые публикации
Синдром Тричер Коллинза
Последняя редакция: 17.10.2021

Весь контент Web2Health проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.

При внутриутробных нарушениях процессов развития костей возникают серьезные черепно-лицевые деформации, и одной из разновидностей такой патологии является синдром Тричер Коллинза (TCS) или мандибулофасциальный, то есть челюстно-лицевой дизостоз.
Код заболевания по МКБ 10: класс XVII (врожденные аномалии, деформации и хромосомные нарушения), Q75.4 - mandibulofacial dysostosis.
Код по МКБ-10
 [
[Причины синдрома Тричер Коллинза
Данный синдром получил имя выдающегося британского офтальмолога Эдварда Тричер Коллинза, описавшего основные черты патологии более ста лет назад. Однако европейские врачи чаще называют этот вид аномалии костей лица и челюстей болезнью или синдромом Франческетти – на основании обширных исследований швейцарского офтальмолога Адольфа Франческетти, который ввел термин «мандибулофасциальный дизостоз» в середине прошлого века. В медицинских кругах также используется название – синдром Франческетти-Коллинза.
Причины синдрома Тричер Коллинза – мутации гена TCOF1 (в локусе хромосомы 5q31.3-33.3), который кодирует ядрышковый фосфопротеин, отвечающий за формирование черепно-лицевой части эмбриона человека. В результате преждевременного уменьшения количества этого белка нарушаются биогенез и функции рРНК. По мнению генетиков исследовательской программы Human Genome, эти процессы приводят к сокращению пролиферации эмбриональных клеток нервного гребня – валика вдоль нервного желоба, который в ходе развития зародыша замыкается в нервную трубку.
Формирование тканей лицевой части черепа происходит благодаря трансформации и дифференциации клеток верхней (головной) части нервного гребня, которые мигрируют вдоль нервной трубки в область первой и второй жаберных дуг зародыша. И дефицит этих клеток вызывает черепно-лицевые деформации. Критический период возникновения аномалий – с 18 по 28 день после оплодотворения. По завершении миграции клеток нервного гребня (на четвертой неделе гестации) образуются практически все рыхлые мезенхимальные ткани в области лица, которые позже (с 5 по 8 недели) дифференцируются в скелетные и соединительные ткани всех частей лица, шеи, гортани, уха (в том числе внутреннего) и будущих зубов.
Патогенез
Патогенез синдрома Тричер Коллинза часто имеет семейный характер, и аномалия наследуется по аутосомно-доминантному принципу, хотя бывают случаи аутосомно-рецессивной передачи дефекта (при мутациях других генов, в частности, POLR1C и POLR1D). Самым непредсказуемым в челюстно-лицевом дизостозе является то, что мутация наследуется детьми только в 40-48% случаев. То есть у 52-60% пациентов причины синдрома Тричер Коллинза не связаны с наличием аномалии в роду, и, как полагают, патология возникает в результате спорадических генных мутаций de novo. Вероятнее всего, новые мутации представляют собой последствия тератогенного воздействия на плод во время беременности.
В числе тератогенных причин данного синдрома специалисты называют большие дозы этанола (этилового спирта), радиацию, сигаретный дым, цитомегавирус и токсоплазму, а также гербициды на основе глифосата (Раундал, Глифор, Торнадо и др.). А в список ятрогенных факторов попали препараты от угрей и себореи с 13-цис-ретиноевой кислотой (Изотретиноин, Аккутан); противосудорожное лекарство Фенитоин (Дилантин, Эпанутин); психотропные средства Диазепам, Валиум, Реланиум, Седуксен.
Симптомы синдрома Тричер Коллинза
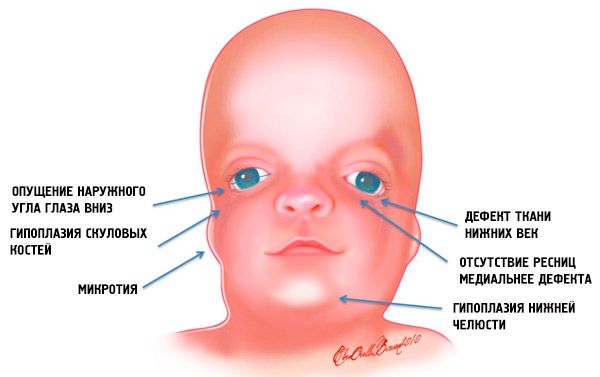
По большей части, клинические признаки мандибулофасциального дизостоза и степень их выраженности зависят от особенностей проявления генных мутаций. И первые признаки данной аномалии в большинстве случаев видны у ребенка сразу же после его появления на свет: лицо при синдроме Тричер Коллинза имеет характерный вид. Причем морфологические аномалии обычно двусторонние и симметричные.
Наиболее очевидные симптомы синдрома Тричер Коллинза:
- недоразвитость (гипоплазия) лицевых костей черепа: скуловых, скуловых отростков лобной кости, боковых крыловидных пластинок, придаточных пазух носа, нижней челюсти и выступов костных эпифизов (мыщелков);
- недоразвитие костей нижней челюсти (микрогнатия) и более тупой чем обычно нижнечелюстной угол;
- нос имеет нормальный размер, однако, кажется большим из-за гипоплазии надбровных дуг и недоразвитости или отсутствия скуловых дуг в области висков;
- глазные щели нисходящие, то есть разрез глаз аномальный, с опущенными вниз наружными уголками;
- дефекты нижних век (колобома) и частичное отсутствие ресниц на них;
- ушные раковины неправильной формы с широким диапазоном отклонений, вплоть до их расположения в углу нижней челюсти, отсутствия мочек, слепых свищей между козелком уха и углом рта и др.;
- сужение или заращение (атрезия) наружного слухового каналов и аномалии косточек среднего уха;
- отсутствие или гипоплазия околоушных слюнных желез;
- фарингеальная гипоплазия (сужение глотки и дыхательных путей);
- несращение твердого нёба (волчья пасть), а также отсутствие, укорочение или неподвижность мягкого неба.
Такие анатомические аномалии во всех случая имеют осложнения. Это функциональные нарушения слуха в виде проводящей (кондуктивной) тугоухости или полной глухоты; нарушения зрения из-за неправильно формирования глазных яблок; дефекты нёба вызывают трудности с кормлением и глотанием. Имеются связанные с дефектами челюстей нарушения окклюзии зубов (неправильный прикус), что, в свою очередь, вызывает проблемы с жеванием и артикуляцией. Патологии мягкого неба объясняют гнусавость голоса.
Осложнения и последствия
Последствия челюстно-лицевых аномалий при синдроме Тричер Коллинза проявляются в том, что при рождении ребенка его интеллектуальные способности нормальные, но из-за дефектов слуха и других нарушений отмечается вторичная задержка умственного развития.
Кроме того, дети с такими дефектами остро чувствуют свою ущербность и страдают, что негативно сказывается на их нервной системе и психике.
Диагностика синдрома Тричер Коллинза
Постнатальная диагностика синдрома Тричер Коллинза, по существу, проводится на основании клинических признаков. Челюстно-лицевой дизостоз легко определяется при полный экспрессивности синдрома, но когда присутствуют минимально выраженные симптомы патологии, с постановкой правильного диагноза могут возникнуть проблемы.
При этом особого внимания требует оценка всех связанных с аномалиями функций, особенно тех, что затрагивают дыхание (в связи с угрозой апноэ во сне). Также проводится оценка и мониторинг эффективности кормления и насыщения гемоглобина кислородом.
В дальнейшем - на 5-6 день после рождения - предстоит выяснить степень повреждений слуха с помощью аудиологического тестирования, которое должно проводиться еще в родильном доме.
Назначается обследование, в ходе которого инструментальная диагностика проводится рентгеноскопией черепно-лицевой дисморфологии; пантомографией (панорамным рентгеном костных структур лицевого черепа); полной черепной компьютерной томографией в различных проекциях; КТ или МРТ головного мозга для определения состояния внутреннего слухового прохода.
Самое раннее – пренатальное – диагностирование челюстно-лицевых аномалий при наличии синдрома Тричер Коллинза в семейном анамнезе возможно путем биопсии ворсин хориона на 10-11 неделе беременности (процедура угрожает выкидышем и занесением инфекции в матку).
Также берутся анализы крови членов семьи; на 16-17 неделе беременности берется анализ околоплодных вод (трансабдоминальный амниоцентез); на 18-20 неделях беременности проводится фетоскопия и берется кровь из плодовых сосудов плаценты.
Но чаще всего в дородовой диагностике данного синдрома у плода используется УЗИ (на 20-24 неделях беременности).
Какие анализы необходимы?
Дифференциальная диагностика
Этими же методами специалисты пользуются, когда нужна дифференциальная диагностика, чтобы распознать неярко выраженный синдром Тричер Коллинза и отличить его от других врожденных аномалий черепно-лицевых костей, в частности: синдромов Апера, Крузона, Нагера, Петерс-Хевельса, Хеллермана-Штефа, а также с гемифациальной микросомии (синдрома Гольденхара), гипертелоризма, преждевременного заращения швов черепа (краниостеноза) или нарушения сращения лицевых костей (краниосиностоза).
Лечение синдрома Тричер Коллинза
Как и во всех случаях, генетически обусловленных врожденных дефектов, лечение синдрома Тричер Коллинза в тяжелых формах носит исключительно паллиативный характер, поскольку терапевтических методов при таких патология просто не существует. Спектр и степень деформаций при данном синдроме обширны и, следовательно, характер и интенсивность врачебного вмешательства также имеет множество вариантов.
Для коррекции и улучшения слуха используются слуховые аппараты, для улучшения речи – занятия с логопедом.
Хирургические вмешательства требуются в раннем возрасте в тяжелых случаях сужения дыхательных путей (проводят трахеостомию) и гортани (выполняется гастростоммия для кормления) Также может потребоваться оперативная коррекция нёба.
Операции по удлинению нижней челюсти выполняются в возрасте 2-3 лет или позже. Реконструкция мягких тканей включает в себя коррекцию колобомы нижнего века и пластику ушных раковин.